Volume 12 | Issue 2 | Apr-June 2017 | Page 27-30 | Rahul Bansal, P B Mohammed Farook, Angad Jolly, Idris Kamran
Authers: Rahul Bansal [1], P B Mohammed Farook [1], Angad Jolly [1], Idris Kamran [1]
[1] Department of Orthopedics Mysore Medical College & Research Institute, Mysore -570001, Karnataka, India.
Address of Correspondence
Dr. Rahul Bansal
Room 101, PG Men’s Hostel MMC&RI, Irwin Road, Mysore – 570001.
Email ID: 85rahulbansal@gmail.com.
Abstract
Background: Pycnodysostosis (PKND) is a rare autosomal recessive disorder. This disorder is classified as a lysosomal storage disorder due to defective gene encoding for Cathepsin K (CTSK). Patients with PKND usually present early with short stature, sclerotic bone which are fragile and are known to cause pathological fractures of long bones. Patients have typical oral, dental and maxillofacial manifestations. These patients are described to have normal intelligence and sexual development. Fracture healing in these patients is debatable [3]. We are describing here a case of 18-year-old male patient with mild mental retardation, conductive deafness and atypical skeletal manifestations such as short metatarsals. But, he had normal clavicle and mandible with no dental abnormality which, is most commonly associated with this condition [4]. Pathological fracture of tibial shaft which was operated and fixed had normal union. The diagnosis was made based on clinical and radiological findings in most cases reported earlier but our diagnosis was confirmed with genetic analysis.
Keywords: Pycnodysostosis, PKND, Cathepsin K, CTSK gene, Sclerotic bone disease, pathological fracture.
Introduction
Pycnodysostosis is also known as osteopetrosis acro-osteolytica or Toulouse-Lautrec syndrome. It was first described by Maroteaux and Lamy in 1962 [1]. It is also known by the name Toulouse-Lautrec syndrome, after the French artist Henri de Toulouse-Lautrec, who was believed to be suffering from the disease [1]. It is a rare autosomal recessive disorder with incidence of 1.7 per 1 million births [2.3,4]. PKND is now mapped to the human chromosome lq21. It is a lysosomal storage disorder with mutation in CTSK gene coding for Cathepsin K which, is lysosomal cysteine protease and is expressed in osteoclasts. Deficiency of lysosomal cysteine protease results in defective bone matrix resorption which leads to sclerotic bone formation [5]. This genetic disorder is linked to consanguinity with no sex predilection, and is usually diagnosed early due to short stature but very commonly diagnosed late usually when patient presents with a pathological fracture of long bones. It can be confused with other sclerotic bone disease like Osteopetrosis (Albers-Schonberg disease), Cleidocranial Dysplasia and Idiopathic Acro-Osteolysis. Patients presenting with PKND have common characteristics — morphologically, they are short statured, peculiar facies, broad prominent forehead with beaked nose, short stubby fingers, proptosis with blue sclera, grooved palate, abnormal dentition with malpositioned teeth, delayed eruption and crowding of teeth. Skeletal abnormalities include generalized increase in bone density, cranial dysplasia with delayed closure of cranial stures, obtuse-angled mandible, shortening of terminal phalanx, dysplastic lateral end of clavicles. Patients most commonly present with pathological fractures of lower limb bones due to increased bone fragility. They have normal intelligence, sexual development and normal life expectancy.
Case Report
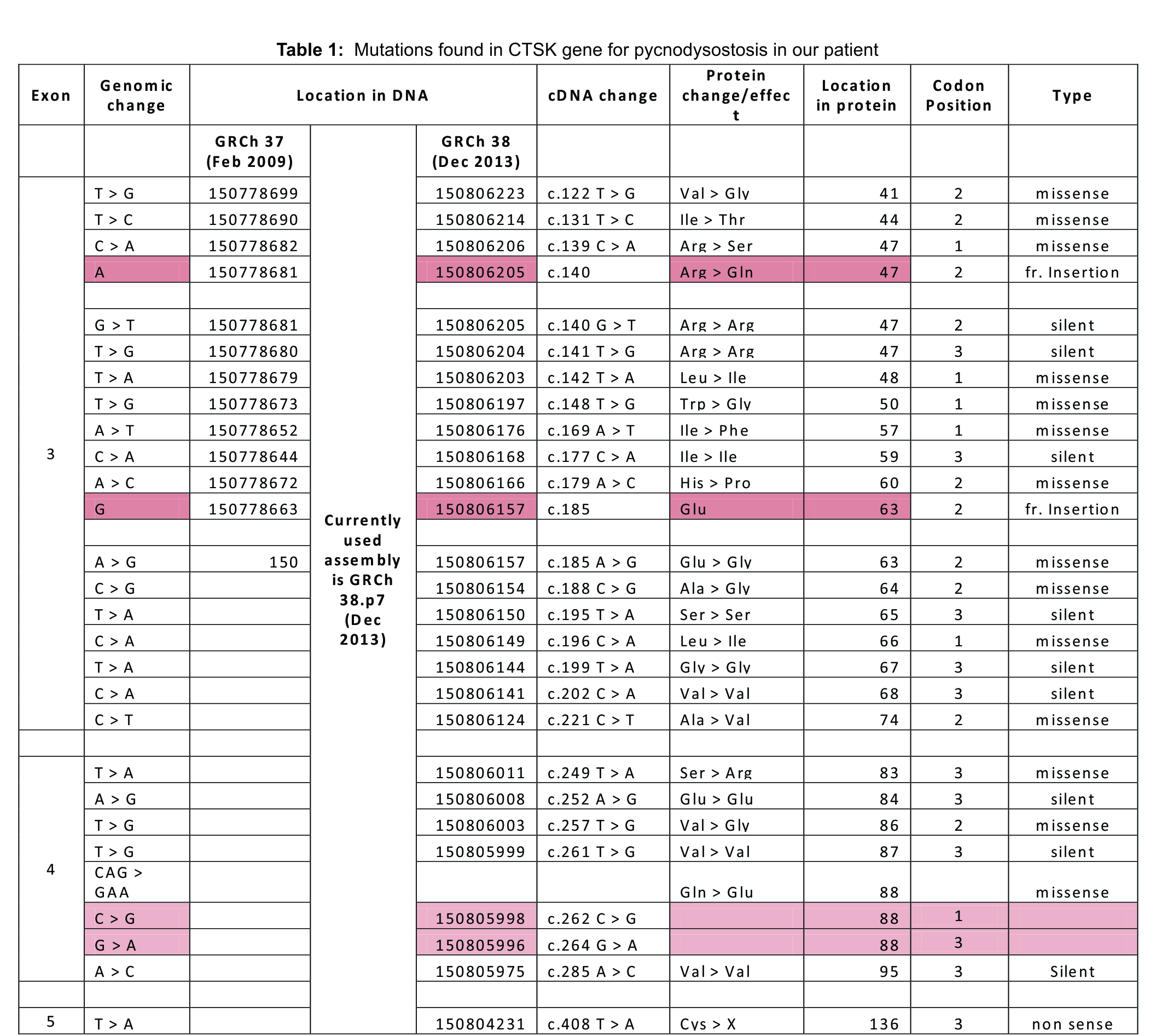
A young male teenager who appeared to be in his early teens was brought to the orthopedic outpatient clinic at our teaching hospital following a trivial fall. He had all the clinical features of a closed diaphyseal fracture of his right tibia. Radiograph confirmed our diagnosis but there seemed to be old united fractures and bones were sclerotic with loss of corticomedullary differentiation which led us to believe that this was a case of Osteopetrosis. On detailed history, we found that patient had similar fractures of tibia of both legs following trivial trauma three times on each side. These were treated conservatively and the fractures had united well. We now realized that patient was 18 years old and he certainly was short for his age. He was deaf from early childhood, so his speech was not well developed either. He was conceived out of a second degree consanguineous marriage. His younger sibling was 12 years old, who was said to be dumb and mentally-retarded, also had left sided motor weakness; sibling could not be examined and was not brought to the hospital after repeated requests. On gross appearance he had facial dysmorphia and coarse facial features with frontal bossing and large forehead, deep set eyes, prominent nose, mandibular prognathism, large tongue, widely spaced nipples (Fig. 1), short neck with low set hairline, protruded chest more prominent on left side (Fig. 1), both hands appeared short and stubby with discolored brittle nails (Fig. 2) and sandal gap deformity bilaterally (Fig. 3). Further evaluation revealed that he had middle ear deafness and ophthalmological examination was normal with normal fundus on either side. His metal age was about 7.5 years, suggesting mild to moderate mental retardation. Anthropometric measurement confirmed wasting, stunting with height of 144cms (Fig. 4), cephalic index of 96.15 suggesting brachycephaly. Radiographs revealed old united fractures in bilateral tibia and a fresh transverse fracture on right (Fig. 5a). They revealed generalized sclerosis of all the bones, acrolysis of terminal phalanx in hands (Fig. 6), short 3rd, 4th and 5th metatarsals (Fig. 7) and open anterior fontanelle (Fig. 8). Mandible appeared normal with normal dentition, chest X-ray was normal with normal clavicle bilaterally and there was no scoliosis or spine deformity. His labs were normal with no features of anemia clinically or in blood investigations. Calcium was borderline at 8.3mg/dl but parathyroid hormone, thyroid profile, liver functions and renal functions were all normal. Based on clinical features and radiographs it became clear to us that we were dealing with a rare case of Pycnodysostosis and not Osteopetrosis. Patient was posted for elective surgery and was operated under general anesthesia with no difficulty in intubation, his intra operative and post-operative periods were uneventful. The fracture was fixed with 9 holed DCP by carefully drilling the bone and taking care of the drill bits (Fig. 5b). Patient was then discharged with a cast and advised not to bear weight; family was offered genetic counselling with precautions to be taken. At the time of discharge we confirmed the diagnosis with genetic testing which is not easily available in our country and affordability is another major factor. We were able to get the genetic test done and the results came positive for CTSK gene mutation. Mutations found in the CTSK gene in our patient were tabulated and recorded (Table. 1). With regular follow up fracture showed good signs of heeling and union was achieved at 3 months with satisfactory radiographs (Fig. 5C). Patient is now bearing weight normally with no difficulty in ambulation. There were no issues such as nonunion, malunion or pseudo arthrosis with any of the fractures of tibia in both lower limbs.

Discussion
Pycnodysostosis is a rare sclerotic bone disease with autosomal recessive inheritance pattern. Genetic defect in this condition is due to mutation in CTSK gene located on chromosome 1q21. CTSK is a lysosomal cysteine protease responsible for osteoclastic activity via degradation of collagen Type I and Type II, osteonectin and osteopontin which, helps in bone resorption and bone remodeling. Bones in this condition appear sclerotic and are abnormally dense, but brittle because of this reduced clastic activity [5]. This increases bone fragility and multiple pathological bone fractures most commonly that of long bones of lower limbs. Patient usually present early with concerns of short stature so, most patients are stunted as in our case. But when they present late, it is because of fragile bones and multiple fractures of bones. These patients have typical coarse facial features with large head (brachycephaly) with frontal bossing , prominent nose with depressed bridge, occasional wide protruding eyes with blue sclera, large protruding tongue, high-arched palate, narrow chest, short stubby hands with dystrophic nails, sandal gap deformity in feet[6]. Spine may show deformities like exaggerated lumbar lordosis or kyphosis and scoliosis but our patient had normal spine on examination [6].
Additionally, our patient also had low set hairline with short neck, widely-spaced nipples and mild pigeon chest deformity on left side but these features have not been reported in older cases. Sexual development is normal with normal life span in these patients but our patient had Tanner stage 4 of development for both genitals as well as pubic hair, with undescended testicle on the right side which has never been reported.
There is generalized bone sclerosis of all the bones on radiographs with, loss of corticomedullary differentiation but medullary canal itself is not obliterated and long bones appear thin and tubular. Most common radiological manifestations in this condition is acro-osteolysis of terminal phalanx of hands [7]. Our patient in addition showed short third, fourth and fifth metatarsals which is an uncommon presentation. Skull shows open anterior fontanelle and cranial sutures, hypoplasia of paranasal sinuses. Dysplastic clavicles are very common feature seen in most cases reported in the past but our patient had normal clavicle and there was no hypoplasia of lateral end as reported in many cases [8]. Osteomyelitis is a common complication in adults which happens due to oral manifestation. Oral manifestations include dental malformations with crowding of teeth or supernumerary teeth, delayed eruption of teeth with caries is common, and mandible is hypoplastic with loss of mandibular angle or obtuse angled mandible. In our patient there were no dental malformations with any mandibular hypoplasia and mandibular angle was maintained [9].
Patient are reported to have difficult intubation due to high arched palate, mandibular hypoplasia and larger tongue, but there were no such issues in our case, and this is an important thing to be considered while posting such patients for surgeries. Such features also pre dispose patients to have obstructive sleep apnea as is the case with our patient. Fracture healing is debatable, textbooks mention normal fracture union but multiple studies show increased rate of malunion, pseudoarthrosis and nonunion [10]. In our case old fractures had healed normally with no malunion and the fracture which was fixed also healed normally. Laboratory investigations are usually within normal limits with normal biochemistry and normal hematological parameters, it has been reported that there may be raised calcitonin levels. Histologically it is similar to Osteopetrosis. PKND can be confused with other conditions based on either external morphological features or sclerotic bones when seen on radiographs. Pycnodysostosis has to be differentiated from Achondroplasia, Osteopetrosis and Cleidocranial dysostosis.
Achondroplasia is a condition where patients have dwarfism with shorter limbs but normal torso and PKND patients on first appearance can be confused with this condition. But achondroplasia patients have normal bone texture, cranial sutures close normally. Proliferation of cartilage is affected at the epiphyseal end plates of the long bones which results in short bones, but the ossification is normal. Osteopetrosis has a dominant inheritance pattern with sclerotic dense bones but patients usually have aplastic anemia as medullary canal is obliterated. Patients here have visceral manifestations like hepatosplenomegaly but there is no phalangeal or clavicular hypoplasia and cranial sutures close normally. Cleidocranial dysostosis has autosomal dominant inheritance with normal stature and no sclerosis of bones but dysplastic or absent one or both clavicles.
Conclusion
Our patient showed morphological feature of short neck, low set hair line, wide spaced nipples and pigeon chest deformity in addition to other usual features which have not been reported earlier. This patient also had undescended testicle on right side and mild to moderate mental retardation. We are not sure if these features were part of PKND or completely unrelated or had any other genetic association as his younger brother is said to have mental retardation. Case presented here had most of the common radiological findings but had absolutely normal clavicle and mandible with normal dentition which is very common feature of PKND reported in most of the cases. In addition, our patient had short third, fourth and fifth metatarsals bilaterally which have not been reported in the past.
We suggest complete examination of such patients with such varied morphological and radiological features to see if these features are part of only PKND or overlap with other inherited genetic conditions.
Fracture healing was normal with no malunion or pseudo arthrosis as reported by many which, is why fracture management is very important in these cases. After fixation or conservative management, it is important to maintain adequate satisfactory reduction with delaying the weight bearing in these patients.
Finally, genetic testing of CTSK gene mutation is the only confirmatory test for this condition and has not been used in earlier cases reported in developing countries. It is an important tool which, if made affordable and easily available would help us diagnose more cases which are left out or confused with other sclerotic bone diseases. We feel that these cases are underdiagnosed and PKND may not be such a rare disease as thought to be.
References
1. Maroteaux P, Lamy M (1962) Pyknodysostosis. Presse Med 70:999-1002.
2. S. M. Krane and A. L. Schiller (1998) Hyperostosis, fibrous dysplasia, and other dysplasias of bone and cartilage. In: Harrison’s Principles of Internal Medicine, A. S. Fauci, E. Braunwald, K. J. Isselbacher et al., Eds. McGraw-Hill, NewYork, pp. 2269–2275.
3. Jiya TU, Hindrik M, Kleipool, Ham J (2006) Diaphyseal femur fracture in pycnodysostosis treated with pennig wrist external fixator: a case study. J Trauma 49(5):477-479.
4. K. W. Fleming, G. Barest, and O. Sakai (2007) Dental and facial bone abnormalities in pyknodysostosis: CT findings. American Journal of Neuroradiology 28.1:132–134.
5. Motyckova G, Fisher DE (2002) Pycnodysostosis: Role and Regulation of Cathepsin K in Osteoclast Function and Human Disease. Curr Mol Med 2:407-421.
6. Kumar R, Misra PK, Singhal R (1988) An unusual case of pycnodysostosis. Arch Dis Child 63:558–9.
7. Wolpowitz A, Matisonn A (1974) A comparative study of pycnodysostosis, cleidocranial dysostosis, osteopetrosis and acro-osteolysis. S Afr Med J 48:1011-1018.
8. Satoru Shiraishi (1971) Pycnodysostosis: (dysostosis petrosans). Acta Orthopaedica Scandinavica 42.3:227-243.
9. S., Rohit et al. (2015) Osteomyelitis in Pycnodysostosis – Report of 2 Clinical Cases. Journal of Clinical and Diagnostic Research 9.1:15–17.
10. Flávio Dorcilo Rabelo, Carlos Henrique Ribeiro do Prado, Flávio Leão Rabelo, Letícia Martins (2010) Reconciderations regarding time of fracture healing in Pycnodysostosis. Rev Bras Ortop 45.6:606-611.
| How to Cite this article: Bansal R, Farook PBM, Jolly A, Kamran I . Pycnodysostosis a case report : Unusual presentation with pathological tibial fracture treated and confirmed with genetic analysis. Journal of Trauma and Orthopaedic Surgery. April – June 2017;12(2):27-30. |